Hemokromatoz ilk dəfə 1889-cu ildə ayrı bir xəstəlik olaraq təsvir edildi. Ancaq xəstəliyin səbəblərini yalnız tibbi genetikanın inkişafı ilə dəqiq müəyyən etmək mümkün oldu.
Belə bir gec təsnifat xəstəliyin təbiəti və onun kifayət qədər məhdud paylanmasına kömək etdi.
Beləliklə, müasir məlumatlara görə, dünya sakinlərinin 0.33% -i hemokromatoz inkişaf riski altındadır. Xəstəliyə səbəb olan nədir və onun əlamətləri nədir?
Hemochromatosis - bu nədir?
 Bu xəstəlik irsi xarakter daşıyır və simptomların çoxluğu və ciddi ağırlaşmaların və əlaqəli patologiyaların yüksək riski ilə xarakterizə olunur.
Bu xəstəlik irsi xarakter daşıyır və simptomların çoxluğu və ciddi ağırlaşmaların və əlaqəli patologiyaların yüksək riski ilə xarakterizə olunur.
Tədqiqatlar göstərir ki, hemokromatoz ən çox HFE genində bir mutasiya səbəb olur.
Bir gen çatışmazlığı nəticəsində, onikibarmaq bağırsaqda dəmirin tutulması mexanizmi pozulur.. Bu, bədənin bədəndə dəmirin olmaması ilə bağlı yalan bir mesaj almasına və aktiv və artıq miqdarda dəmiri bağlayan xüsusi bir zülal sintez etməsinə səbəb olur.
Bu, daxili orqanlarda hemosiderinin (bezli piqment) həddindən artıq çökməsinə səbəb olur. Protein sintezinin artması ilə yanaşı, mədə-bağırsaq traktının aktivləşməsi baş verir, bu da bağırsaqdakı qidadan dəmirin həddindən artıq udulmasına səbəb olur.
Beləliklə, normal qidalanma halında da bədəndə olan dəmir miqdarı normadan dəfələrlə çoxdur. Bu, daxili orqanların toxumalarının məhvinə, endokrin sistemlə bağlı problemlərə və toxunulmazlığa səbəb olur.
Növləri, formaları və mərhələləri üzrə təsnifatı
Tibbi praktikada xəstəliyin ilkin və ikinci dərəcəli növləri bölünür. Bu vəziyyətdə, irsi olaraq da adlandırılan ibtidai, bir genetik meylin nəticəsidir. İkinci dərəcəli hemokromatoz, vəzi mübadiləsində iştirak edən ferment sistemlərinin işində sapmaların inkişafının nəticəsidir.
İrsi (genetik) xəstəliyin dörd forması məlumdur:
- klassik
- yetkinlik yaşına çatmayan;
- irsi HFE-əlaqəsiz növlər;
- otosomal dominant.
Birinci növ, altıncı xromosoma bölgəsinin klassik resessiv mutasiyası ilə əlaqələndirilir. Bu tip halların böyük əksəriyyətində diaqnoz qoyulur - xəstələrin 95 faizindən çoxu klassik hemokromatozdan əziyyət çəkir.
Yetkinlik yaşına çatmayan xəstəlik növü, başqa bir gen, HAMPdakı bir mutasiya nəticəsində meydana gəlir. Bu dəyişikliyin təsiri ilə orqanlarda dəmir çöküntüsündən cavabdeh olan bir ferment olan hepcidinin sintezi əhəmiyyətli dərəcədə artır. Adətən xəstəlik on-otuz yaş arasında özünü göstərir.
HFE-əlaqəsiz növü HJV geninin uğursuz olduğu zaman inkişaf edir. Bu patoloji transferrin-2 reseptorlarının hiperaktivasiya mexanizmini əhatə edir. Nəticədə hepcidin istehsalı intensivləşir. Yetkinlik yaşına çatmayan xəstəlik növü ilə fərq, ilk vəziyyətdə dəmir bağlayan fermentin istehsalına birbaşa cavabdeh olan bir genin uğursuz olmasıdır.
 Halbuki ikinci vəziyyətdə, orqanizm fermentin istehsalına səbəb olan qida içərisində dəmirin çox olması vəziyyətini yaradır.
Halbuki ikinci vəziyyətdə, orqanizm fermentin istehsalına səbəb olan qida içərisində dəmirin çox olması vəziyyətini yaradır.
İrsi hemokromatozun dördüncü növü SLC40A1 geninin nasazlığı ilə əlaqələndirilir.
Xəstəlik qocalıqda özünü göstərir və dəmir birləşmələrinin hüceyrələrə daşınması üçün cavabdeh olan ferroportin proteininin qeyri-sintezi ilə əlaqələndirilir.
Missens mutasiya səbəbləri və risk faktorları
 Xəstəliyin irsi tipində genetik mutasiya bir insanın meylinin nəticəsidir.
Xəstəliyin irsi tipində genetik mutasiya bir insanın meylinin nəticəsidir.
Tədqiqatlar göstərir ki, xəstələrin əksəriyyəti Şimali Amerika və Avropanın ağ sakinləridir, İrlandiyadan gələn immiqrantlar arasında ən çox sayda hemokromatozdan əziyyət çəkən insanlardır.
Üstəlik, fərqli mutasiyaların yayılması dünyanın müxtəlif bölgələri üçün xarakterikdir. Kişilər qadınlara nisbətən xəstəliyə bir neçə dəfə daha həssasdırlar. Sonuncu, simptomlar ümumiyyətlə menopoz zamanı bədəndəki hormonal dəyişikliklərdən sonra inkişaf edir.
Qeydiyyata alınan xəstələr arasında qadınlar kişilərə nisbətən 7-10 dəfə azdır. Dəyişmənin səbəbləri hələ də bəlli deyil. Xəstəliyin yalnız irsi təbiəti təkzibolunmaz şəkildə sübut edilir və hemokromatoz ilə qaraciyər fibrozunun mövcudluğu arasındakı əlaqə də izlənilir.
 Birləşdirici toxumanın böyüməsini orqanizmdə dəmirin yığılması ilə izah etmək mümkün olmasa da, hemokromatozlu xəstələrin 70% -də qaraciyər fibrozu var.
Birləşdirici toxumanın böyüməsini orqanizmdə dəmirin yığılması ilə izah etmək mümkün olmasa da, hemokromatozlu xəstələrin 70% -də qaraciyər fibrozu var.
Üstəlik, bir genetik meyl mütləq xəstəliyin inkişafına səbəb olmur.
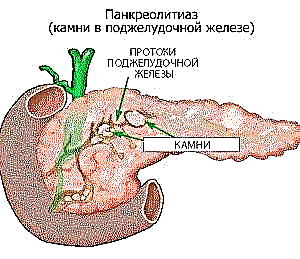
Bundan əlavə, ilkin normal genetikası olan insanlarda müşahidə olunan hemoqromatozun ikinci dərəcəli forması var. Risk faktorlarına bəzi patologiyalar da daxildir. Beləliklə, köçürülmüş steatohepatit (adipoz toxumasının alkoqolsuz çökməsi), müxtəlif etiologiyalı xroniki hepatitlərin inkişafı, həmçinin mədəaltı vəzinin tıxanması xəstəliyin təzahürünə kömək edir.
Qadınlarda və kişilərdə hemokromatozun simptomları
 Əvvəllər yalnız bir sıra ciddi simptomatik təzahürlərin inkişafı bu xəstəliyin diaqnozu qoyulmasına imkan verdi.
Əvvəllər yalnız bir sıra ciddi simptomatik təzahürlərin inkişafı bu xəstəliyin diaqnozu qoyulmasına imkan verdi.
Həddindən artıq dəmir yığan xəstə xroniki yorğunluq, zəiflik hiss edir.
Bu simptom hematokromatozu olan xəstələrin 75% -i üçün xarakterikdir. Dəri piqmentasiyası inkişaf etdirilir və bu proses melanin istehsalı ilə əlaqələndirilmir. Orada dəmir birləşmələrinin yığılması səbəbindən dəri daha qaranlıq olur. Qaranlıq xəstələrin 70% -dən çoxunda müşahidə olunur.
Yığılmış dəmirin immun hüceyrələrinə mənfi təsiri toxunulmazlığın zəifləməsinə səbəb olur. Buna görə xəstəliyin gedişi ilə xəstənin infeksiyalara həssaslığı artır - normal şəraitdə olduqca ciddi və zərərsizdir.
 Xəstələrin təxminən yarısı ağrının meydana gəlməsində ifadə olunan birgə patologiyalardan əziyyət çəkirlər.
Xəstələrin təxminən yarısı ağrının meydana gəlməsində ifadə olunan birgə patologiyalardan əziyyət çəkirlər.
Onların hərəkətliliyində də pisləşmə var. Bu simptom meydana gəlir, çünki dəmir birləşmələrinin çox olması oynaqlarda kalsium yataqlarını katalizləşdirir.
Aritmiya və ürək çatışmazlığının inkişafı da mümkündür. Mədəaltı vəzinə mənfi təsir tez-tez şəkər xəstəliyinə səbəb olur. Aşırı dəmir tər vəzilərinin disfunksiyasına səbəb olur. Nadir hallarda baş ağrısı müşahidə olunur.
Xəstəliyin inkişafı kişilərdə iktidarsızlığa səbəb olur. Azaldılmış cinsi funksiya, bədənin dəmir birləşmələri məhsulları ilə zəhərlənmə əlamətlərini göstərir. Qadınlarda tənzimləmə zamanı ağır qanaxma mümkündür.
 Mühüm bir simptom, qaraciyərdə artım, həmçinin olduqca güclü qarın ağrısıdır, görünüşü ilə sistematik olaraq müəyyənləşdirmək mümkün deyil.
Mühüm bir simptom, qaraciyərdə artım, həmçinin olduqca güclü qarın ağrısıdır, görünüşü ilə sistematik olaraq müəyyənləşdirmək mümkün deyil.
Bir neçə simptomun olması xəstəliyin dəqiq laborator müayinəsinə ehtiyac olduğunu göstərir.
Xəstəliyin əlaməti qan içində yüksək hemoglobin tərkiblidir, eyni zamanda az miqdarda qırmızı qan hüceyrələrində olur. Transferrin dəmir ilə 50% -dən aşağı doyma göstəriciləri hemokromatozun laborator əlaməti hesab olunur.
Dokularının yüksək bir sıxlığı olan qaraciyərdə əhəmiyyətli bir artım da xəstəliyin əlamətidir. Bundan əlavə, hemokromatozla qaraciyər toxumasının rəngində dəyişiklik müşahidə olunur.
Uşaqda necə özünü göstərir?
 Erkən hemokromatozun bir sıra xüsusiyyətləri var - buna səbəb olan mutasiyalardan müvafiq xromosoma bölgələrinə qədər xarakterik klinik mənzərə və təzahürlərə qədər.
Erkən hemokromatozun bir sıra xüsusiyyətləri var - buna səbəb olan mutasiyalardan müvafiq xromosoma bölgələrinə qədər xarakterik klinik mənzərə və təzahürlərə qədər.
Əvvəla, erkən yaşda xəstəliyin əlamətləri polimorfikdir.
Uşaqlar portal hipertenziyanı göstərən simptomların inkişafı ilə xarakterizə olunur. Yeməyin assimilyasiyasının pozulmasını, dalağın və qaraciyərin eyni vaxtda artmasını inkişaf etdirir.
Patoloji inkişafı ilə, ağır və müalicəvi təsirlərə qarşı davamlı astsitlər - qarın bölgəsində meydana gələn damcı başlayır. Özofagusun varikoz damarlarının inkişafı xarakterikdir.
Hansı testlər və diaqnostik metodlar patologiyanı təyin etməyə kömək edir?
 Xəstəliyi müəyyən etmək üçün bir neçə fərqli laboratoriya diaqnostik üsulu istifadə olunur.
Xəstəliyi müəyyən etmək üçün bir neçə fərqli laboratoriya diaqnostik üsulu istifadə olunur.
Əvvəlcə qan nümunəsi qırmızı qan hüceyrələrində və plazmadakı hemoglobinin səviyyəsini öyrənmək üçün edilir.
Dəmir metabolizmasının qiymətləndirilməsi də aparılır.
Desferal test diaqnozu təsdiqləməyə kömək edir. Bunun üçün bir vəzi dərmanının enjeksiyonu aparılır və beş saatdan sonra sidik nümunəsi alınır. Bundan əlavə, daxili orqanların CT və MHİ, onların patoloji dəyişikliklərini - ölçüdə bir artım, piqmentasiya və toxuma quruluşunda dəyişiklik təyin etmək üçün aparılır.
Molekulyar genetik tarama xromosomun zədələnmiş hissəsinin varlığını təyin etməyə imkan verir. Xəstənin ailə üzvləri arasında aparılan bu tədqiqat, xəstəliyi xəstəni narahat edən klinik təzahürləri başlamazdan əvvəl də meydana çıxma ehtimalını qiymətləndirməyə imkan verir.
Müalicə prinsipləri
Müalicənin əsas üsulları bədəndəki dəmir tərkibinin göstəricilərinin normallaşdırılması və daxili orqanlara və sistemlərə ziyan verilməməsidir. Təəssüf ki, müasir tibb gen aparatını necə normallaşdıracağını bilmir.

Qan tökülməsi
Ümumi bir müalicə üsulu qan tökülməsidir. İlkin terapiya ilə həftədə 500 mq qan çıxarılır. Dəmir tərkibinin normallaşmasından sonra, hər üç ayda bir qan nümunəsi meydana gəldikdə, qulluq terapiyasına keçirlər.
Dəmir bağlayıcı dərmanların venadaxili tətbiqi də tətbiq olunur. Beləliklə, aldadıcılar sidik və ya nəcis olan artıq maddələri çıxarmağa imkan verir. Bununla birlikdə, qısa bir fəaliyyət dövrü lazımi xüsusi nasosların köməyi ilə dərmanların müntəzəm dərialtı enjeksiyonunu edir.
Mümkün ağırlaşmalar və proqnoz
Erkən diaqnoz ilə xəstəlik effektiv şəkildə idarə oluna bilər.Daimi qayğı alan xəstələrin həyat müddəti və keyfiyyəti praktik olaraq sağlam insanların həyatından fərqlənmir.
Üstəlik, vaxtında müalicə edilməməsi ciddi fəsadlara yol açır. Bunlara siroz və qaraciyər çatışmazlığı, diabet, qanaxmaya qədər damarlara zərər daxildir.
Kardiyomiyopatiya və qaraciyər xərçənginin inkişaf riski yüksəkdir, intercurrent infeksiyalar da müşahidə olunur.
Oxşar videolar
Hemokromatozun nədir və necə müalicə ediləcəyi haqqında "Sağlam yaşa!" Telekanalında Elena Malysheva ilə: